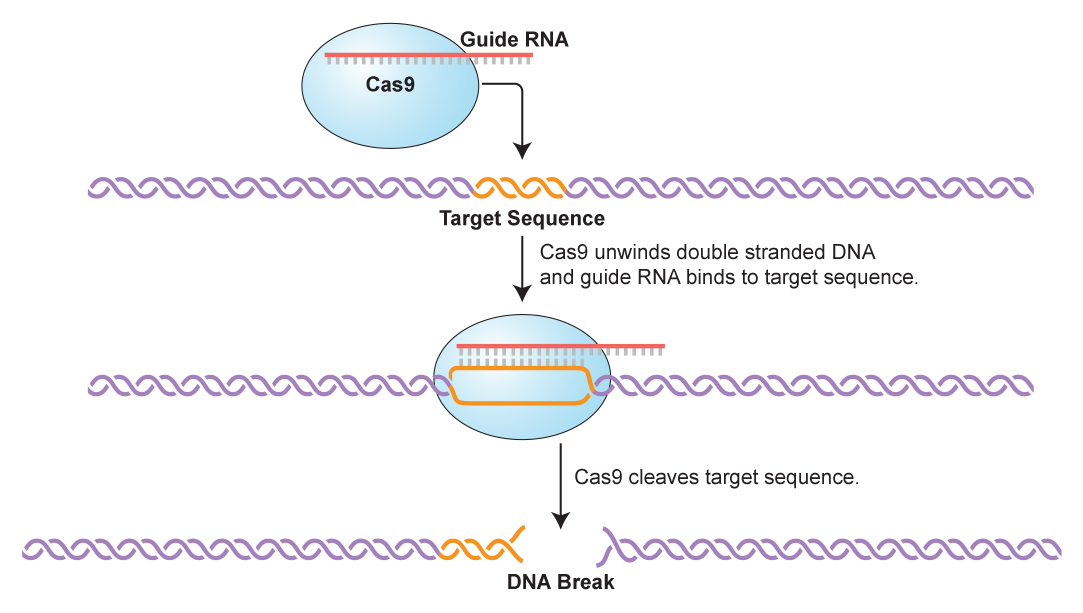
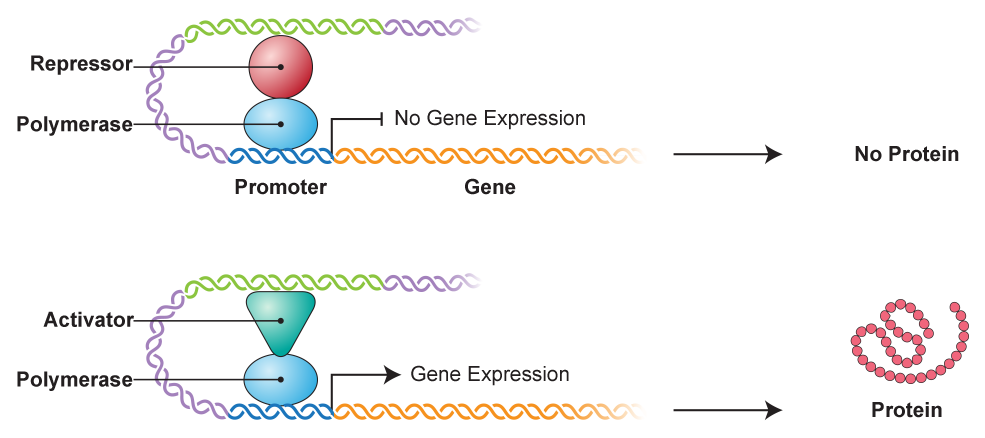
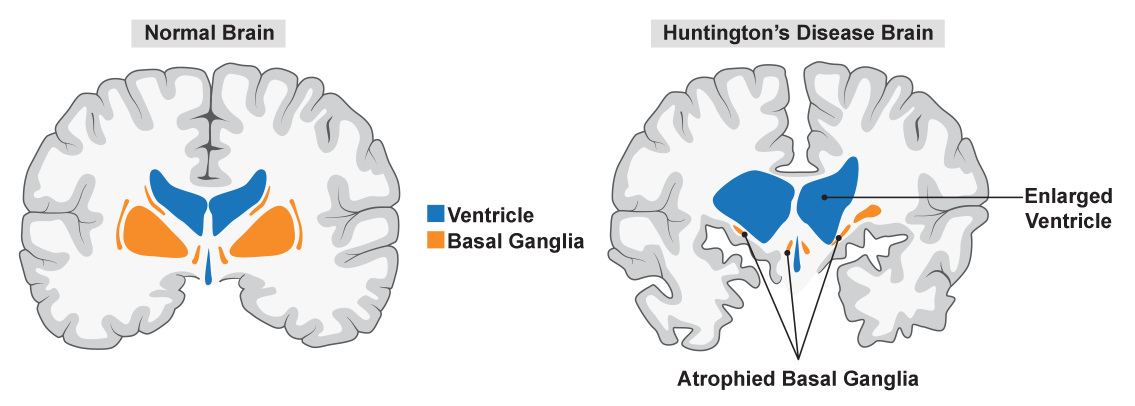
The central dogma of molecular biology is a fundamental concept
in the study of biology, found in all major biology curricula
(NGSS, non-NGSS state-level life science, AP, IB, and
undergraduate Vision and Change) and biology textbooks.
Educators can therefore use this interactive in a variety of
courses, ranging from first-year high school biology to
undergraduate molecular biology electives. The connection to
genetic techniques and their applications extends its use to
biotechnology-focused courses.
Exploration of this interactive ties to several science practices including the use of models and representations, engagement in scientific questioning, construction of evidence-based explanations, and relating knowledge across scales and concepts. The interactive will also emphasize key crosscutting concepts that help students better understand core ideas in science including the importance of structure and function, scale, and cause and effect.
Educators should keep in mind that the flow of information from
DNA to RNA to protein is a dynamic process that involves many
more steps and molecules than are shown in this interactive. In
addition, there are exceptions to this orderly flow. For
example, scientists have discovered that noncoding RNA and
retroviruses are capable of reverse transcription, in which
genetic information flows from RNA to DNA. Educators might also
remind students that, except for cancer, diseases featured in
this interactive are rare monogenic diseases, caused by
mutations in single genes. It will be more challenging to apply
these technologies to more common diseases caused by a
combination of genes and environmental factors. In most cases,
more than one treatment strategy or technology may be applied to
any particular disease.
References
CRISPR
Musunuru, Kiran, Sarah A. Grandinette, Xiao Wang, Taylor R. Hudson, Kevin Briseno, Anne Marie Berry, Julia L. Hacker, et al. 2025. “Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease.” New England Journal of Medicine 392, 22: 2235–2243. https://doi.org/10.1056/NEJMoa2504747.
Leber Congenital Amaurosis
Bainbridge, James W. B., Alexander J. Smith, Susie S. Barker, Scott Robbie, Robert Henderson, Kamaljit Balaggan, Ananth Viswanathan, et al. 2008. “Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis.” New England Journal of Medicine 358, 21: 2231–2239. https://doi.org/10.1056/NEJMoa0802268.
Chacon-Camacho, Oscar Francisco, and Juan Carlos Zenteno. 2015. “Review and Update on the Molecular Basis of Leber Congenital Amaurosis.” World Journal of Clinical Cases 3, 2: 112–124. https://doi.org/10.12998/wjcc.v3.i2.112.
Sickle Cell
Brendel, Christian, Swaroopa Guda, Raffaele Renella, Daniel E. Bauer, Matthew C. Canver, Young Jo Kim, Matthew M. Heeney, et al. 2016. “Lineage Specific BCL11A Knockdown Circumvents Toxicities and Reverses Sickle Phenotype.” Journal of Clinical Investigation 126, 10: 3868–3878. https://doi.org/10.1172/JCI87885.
Frangoul, Haydar, David Altshuler, M. Domenica Cappellini, Yi Shan Chen, Jennifer Domm, Brenda K. Eustace, Juergen Foell, et al. 2021. “CRISPR Cas9 Gene Editing for Sickle Cell Disease and β Thalassemia.” New England Journal of Medicine 384, 3: 252–260. https://doi.org/10.1056/NEJMoa2031054.
Orkin, Stuart H. 2025. “The Fetal to Adult Hemoglobin Switch — Mechanism and Therapy.” New England Journal of Medicine 392, 21: 2135–2149. https://doi.org/10.1056/NEJMra2405260.
Huntington's Disease
Ionis Pharmaceuticals. 2015. “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ISIS 443139 in Patients With Early Manifest Huntington’s Disease.” Clinical trial registration NCT02519036. https://clinicaltrials.gov/ct2/show/NCT02519036.
Kordasiewicz, Holly B., Lisa M. Stanek, Edward V. Wancewicz, Curt Mazur, Melissa M. McAlonis, Kimberly A. Pytel, Jonathan W. Artates, et al. 2012. “Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis.” Neuron 74, 6: 1031–1044. https://doi.org/10.1016/j.neuron.2012.05.009.
Rawlins, Michael D., Nancy S. Wexler, Alice R. Wexler, Sarah J. Tabrizi, Ian Douglas, Stephen J. W. Evans, Liam Smeeth, et al. 2016. “The Prevalence of Huntington’s Disease.” Neuroepidemiology 46, 2: 144–153. https://doi.org/10.1159/000443738.
Duchenne Muscular Dystrophy
Emery, Alan E. H. 1991. “Population Frequencies of Inherited Neuromuscular Diseases—A World Survey.” Neuromuscular Disorders 1, 1: 19–29. https://doi.org/10.1016/0960-8966(91)90039-u.
Kinali, Maria, Virginia Arechavala-Gomeza, Lucy Feng, Sebahattin Cirak, David Hunt, Carl Adkin, Michela Guglieri, et al. 2009. “Local Restoration of Dystrophin Expression with the Morpholino Oligomer AVI 4658 in Duchenne Muscular Dystrophy: A Single-Blind, Placebo-Controlled, Dose-Escalation, Proof-of-Concept Study.” Lancet Neurology 8, 10: 918–928. https://doi.org/10.1016/S1474-4422(09)70211-X.
Cystic Fibrosis
Kosorok, Michael R., Wen-Hsiang Wei, and Philip M. Farrell. 1996. “The Incidence of Cystic Fibrosis.” Statistics in Medicine 15, 5: 449–462. https://doi.org/10.1002/(SICI)1097-0258(19960315)15:5<449::AID-SIM173>3.0.CO;2-X.
Kuk, Kelly, and Jennifer L. Taylor-Cousar. 2015. “Lumacaftor and Ivacaftor in the Management of Patients with Cystic Fibrosis: Current Evidence and Future Prospects.” Therapeutic Advances in Respiratory Disease 9, 6: 313–326. https://doi.org/10.1177/1753465815601934.